Dr. Paul Volden recently presented a webinar on humanized immune system mouse models, focusing on engraftment of PBMCs and isolated cell subsets. Taconic has extensive experience with humanized mice, both from internal research as well as many collaborations with external partners. Although hematopoietic stem cell-engrafted models such as the huNOG and huNOG-EXL may be better known, these alternate humanization approaches offer advantages for certain studies.
We present the full webinar Q&A here.
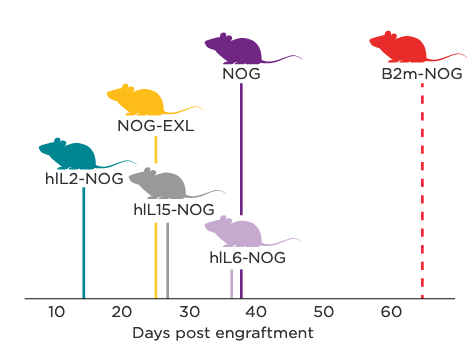
A: We have not worked with NOG mice expressing both hIL-15 and hIL-2, but our colleagues from the Central Institute of Experimental Animals have made this model. It played a minor role in one of their recent publications titled Long-term maintenance of peripheral blood derived human NK cells in a novel human IL-15- transgenic NOG mouse.
 Read the Related Taconic Biosciences' Insights:
Read the Related Taconic Biosciences' Insights:
 Download the Related Taconic Biosciences' White Papers:
Download the Related Taconic Biosciences' White Papers:
We present the full webinar Q&A here.
Graft-vs-Host Disease
Q. What is graft vs. host disease (GvHD) and how is GvHD relevant to mouse models with human immune systems (HIS)?
HSC-engrafted models
Q. HSC-engrafted models require 10-12 weeks for human CD34+ stem cells to engraft and differentiate. Why doesn't this model develop graft vs. host disease during that time?
Q. What humanization percentage is obtained in mice after engraftment with human HSC cells?
PBMC engraftment
Q. Can PBMCs be injected via the intraperitoneal (ip) route or is intravenous (iv) injection always better?
Q. While many studies use iv systemic injection of PBMCs, some studies utilize direct injection of human PBMCs into an engrafted tumor. Is one approach better to use depending on circumstances?
Q. Are there ways to enhance PBMC engraftment?
Graphical representation of terminal-stage GvHD onset in NOG Portfolio models following human PBMC engraftment.
Q. Please elaborate on the benefits of banking PBMCs and other human cells for engraftment. Other than variability concerns, isn't it a good idea to use cells from various different donors?
Other HIS Model Applications
Q. The research reviewed in the webinar covered biologics. Can these models be used to study small molecule drugs?
Q. Transgenic expression of human cytokines in base immunodeficient models is one approach to supporting engraftment and differentiation of particular human immune cell types. Hydrodynamic gene delivery is another approach to get expression of human cytokines in host strains. What are the pros and cons of each approach?
Q. Are there any models that weren't mentioned in the webinar which display good reconstitution of human T cells and human NK cells?
Q. Are humanized mice leveraged that use immune cells coming from different clinical conditions?
Q. How do sex differences in the engrafted cells or across the engrafted host impact humanization?
A: We have not worked with NOG mice expressing both hIL-15 and hIL-2, but our colleagues from the Central Institute of Experimental Animals have made this model. It played a minor role in one of their recent publications titled Long-term maintenance of peripheral blood derived human NK cells in a novel human IL-15- transgenic NOG mouse.
Read the Related Taconic Biosciences' Insights: Download the Related Taconic Biosciences' White Papers: