 Metabolic Dysfunction-Associated Steatohepatitis (MASH) is associated with a chronic proinflammatory state of the liver1-3,10. The clinical pathogenesis of MASH progresses from a steatotic/intralobular inflammatory/hepatocellular ballooning state to progressing levels of fibrosis and in later stages, an increased incidence of cirrhosis and/or hepatocellular carcinoma8-13. The etiology underlying this progression remains unknown, nor are there currently any approved therapies other than lifestyle modifications aimed at modulating or mitigating MASH11,12.
Metabolic Dysfunction-Associated Steatohepatitis (MASH) is associated with a chronic proinflammatory state of the liver1-3,10. The clinical pathogenesis of MASH progresses from a steatotic/intralobular inflammatory/hepatocellular ballooning state to progressing levels of fibrosis and in later stages, an increased incidence of cirrhosis and/or hepatocellular carcinoma8-13. The etiology underlying this progression remains unknown, nor are there currently any approved therapies other than lifestyle modifications aimed at modulating or mitigating MASH11,12.
Identifying New MASH Drug Targets
Han and colleagues sought to identify and define a key pathway responsible for the chronic activation of hepatic proinflammatory immune responses that define the MASH-associated hepatic environment. By understanding the regulators of this immune homeostatic pathway, the authors identify targets that may offer therapeutic potential against MASH.
Macrophages and MASH
The critical role of macrophages in the hepatic proinflammatory state of MASH, specifically Kupffer cells (KC), has supported further investigation into the pathways that regulate their function1,2,4,5,7,8. Macrophages have two functional states: an M1 proinflammatory and a M2 anti-inflammatory polarity1,2,8. The M1 polarized macrophages exacerbate the MASH inflammatory state and result in liver injury via the production of proinflammatory cytokines such as tumor necrosis factor α (TNFα). In contrast, M2 macrophages maintain an anti-inflammatory state by suppression of M1 macrophage activation, apoptosis of M1 macrophages, and the production of anti-inflammatory cytokines such as IL-101,2,6.
The M1/M2 Polarity Switch
The M1/M2 polarization of macrophages is associated with the progression of MASH with patients showing higher M2 levels with steatosis vs. patients with severe steatohepatitis1. Recent studies have shown that by inducing a polarity switch in hepatic macrophages from M1 to M2, it can resolve a high fat diet-induced animal model of MASH1,2,4,5.
Based on their previous work, the authors focused on understanding the pathway governing the polarity switch of M1 to M2 in hepatic macrophages, specifically the involvement of the nuclear receptor retinoic-acid-related orphan receptor α (RORα; NR1F1) in KCs. RORα is a ligand-dependent transcriptional factor that controls an array of target genes associated with both lipid metabolism and inflammation1,2.
ROR Activation in Animal Models
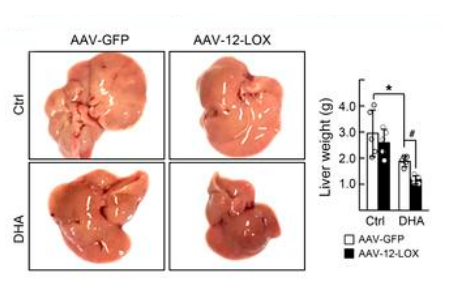
Depicted are a representative sample of the livers and corresponding
liver weights of male C57BL/6 mice exposed to HFD for 16 weeks. Animals
were given either AAV-GFP control or AAV-LOX-12 (5x106 vg; i.v.)
at approximately 13 weeks of diet exposure and were then subsequently
treated for the remaining 3 weeks with DHA (1mg/kg; i.p.; q.d.) or vehicle.
Source: Han et al. JCI 20191
Both patients and animal models of Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) show decreased hepatic expression levels of RORα compared to controls and, furthermore, RORα activation induced a M1 to M2 polarity ratio switch in KCs both in vitro and in vivo1,2.
MASH pathogenesis was exacerbated in a RORα Knockout (KO) mouse model. The RORα KO showed significant alterations in polarity switching with M1 macrophage levels elevated compared to M2. This RORα- dependent exacerbation of MASH was due to effects on the macrophage polarity ratio and not due to alterations in overall macrophage populations1.
Further Investigation of RORα in MASH
Due to the newfound importance of RORα in the development of MASH, the authors sought to identify the components of hepatic milieu that effect the function of this receptor. They narrowed their focus to polyunsaturated fatty acids (PUFAs) and their metabolites due to the altered ratio of PUFAs to saturated fatty acids (high saturated and low PUFAs) in MASH patients1,13. Additionally, FAs are also known to modulate macrophage polarity switch and are thought to be an essential source of M2 activation with docosahexaenoic acid (DHA) being an ideal candidate1.
The authors determined that a metabolite of DHA, maresin-1 (MaR1; unlike DHA) was an endogenous agonistic ligand for RORα in hepatic macrophages.
In Vitro Studies of RORα
In vitro studies showed that MaR1 activation of RORα resulted in a M2 polarity switch (determined via M1/M2 cell marker ratios and M2 signature gene expression levels) in liver macrophages that was both dose and time-dependent1. MaR1 also increased the expression (both mRNA and protein levels) of RORα in liver macrophages1. The effects of MaR1 to induce a M1 to M2 polarity switch in hepatic macrophages required the presence of RORα1.
Further In Vivo Studies
Additionally, in vivo studies showed that Mar1 could inhibit the progression of MASH in an HFD mouse model only in the presence of RORα1. Mar1 increased the number of M2 hepatic macrophages in HFD fed flox control mice, whereas, the levels were unaltered in the HFD fed RORαKO mice1. Similar to RORα, Mar1 expression levels were significantly depressed in both an HFD and methionine choline-deficient (MCD) diet exposed mouse models when compared to controls1. Restoration of Mar1 levels could be addressed via DHA administration but only in the presence of RORα.
Furthermore, clarity was gained on the feedback between Mar1 and RORα when a synthetic agonist to RORα increased Mar1 levels, while a KO of RORα decreased them. The function of RORα is associated with the levels of Mar1 via the effects of RORα on the enzyme 12-lipoxygenase (12-LOX), which is involved in the biosynthesis of Mar11.
Han and colleagues showed that RORα increases levels of Mar1 through induction in the expression of 12-LOX. In vitro work showed hepatic macrophages had significantly elevated levels of 12-LOX when compared to other types of macrophages or hepatocytes. Furthermore, in vivo studies demonstrated that RORαKO mice had reduced levels of 12-LOX compared to control. Activation of RORα via synthetic agonist, endogenous agonist, or infection of AAV-RORα increased 12-LOX levels whereas knockdown of RORα decreased them suggesting 12-LOX is a downstream target of RORα.
Regulating the Protective Effects of DHA
 to anti-inflammatory (M2) phenotype.")
A schematic of the MaR1-RORα-12-LOX autoregulatory circuit. Targeting of this
circuit could provide a therapeutic response in MASH via its resulting polarity
switch in liver macrophages from an inflammatory (M1) to anti-inflammatory (M2)
phenotype. Source: Han et al. JCI 20191
The authors next assessed whether this downstream target of RORα, 12-LOX, could alter the protective effect of DHA administration on an HFD MASH model1. A known inhibitor of 12-LOX (baicalein) abolished the beneficial effects of DHA administration on MASH progression with both hepatic Mar1 levels and M2 macrophages decreasing with coadministration of the inhibitor and DHA.
In contrast, the symptoms of HFD MASH mice significantly improved when given AAV-12-LOX and DHA (reduction of proinflammatory cytokines, lipids, and histopathological progression) with Mar1 and M2 macrophages levels elevated compared to controls1. The potential translation of 12-LOX as a downstream target extended to the clinic, with mRNA levels of 12-LOX significantly lower in MASH patients compared to healthy controls1. Furthermore, RORα and 12-LOX expression levels in humans show a positive correlation, both are decreased in hepatic macrophages of patients with liver fibrosis compared to controls and are induced by Mar1 or a synthetic agonist to RORα in vitro1.
Relevance for MASH Drug Development
Han and colleagues provide evidence of an activation circuit for hepatic macrophage polarity M1 to M2 switch governed by MaR1/RORα/12-LOX which was essential to the beneficial effects of DHA in a mouse model of MASH with partial clinical translation1. A therapeutic approach leveraging a combination of Mar1 and 12-LOX could target this autoregulatory circuit and skew the polarity of liver macrophages providing beneficial anti-inflammatory effects and potential slowing of MASH progression in the clinic.



