Laura Griffin, PhD & Michael Carr
Preclinical Models to Support Newly Approved Therapy for ALS: The SOD1 Rat
Friday, September 15th, 2023

Amyotrophic lateral sclerosis (ALS) is a rare neurological disorder characterized by motor neuron degeneration in the primary motor cortex, brainstem, and spinal cord, which results in progressive muscular atrophy, weakness, and fatality1. Onset of this disease typically occurs in adulthood, and early symptoms are difficult to recognize given their similarity to other neurological disorders2. Incidence is most likely to occur between the ages of 60 and 79 years, and men tend to have higher risk than women (3 per 100,000 person-years vs 2.4 per 100,000 person-years)3.
ALS is characterized as either sporadic or familial. Sporadic ALS occurs in individuals with no family history of the disease and comprises 90-95% of all cases. Familial ALS comprises the remaining 5-10% of all cases and is classified as occurring in at least two people in the same family. Multiple genetic mutations have been implicated in familial ALS but have also been identified in people with sporadic ALS. Exact pathogenesis is unclear, but it is generally accepted that both genetic and non-genetic factors play a role in disease onset.
Since the early 1990s, mutations in more than 40 genes have been implicated in ALS onset. Mutations of the SOD1 gene, which encodes the essential antioxidant enzyme, Cu/Zn superoxide dismutase, were the first to be discovered and linked to familial ALS. The clinical phenotype of ALS patients demonstrating SOD1 mutations is highly variable with respect to symptoms and speed of disease progression, and it is believed that the pathogenicity of mutations is due to accumulation of misfolded protein aggregates, rather than a lack of functionality4. Until recently, there were no approved and available treatments targeting a genetic mutation associated with ALS.
SOD1 in the News
In early 2023, Ionis Pharmaceuticals applied for accelerated approval of tofersen, an investigational antisense medicine that has shown efficacy in people with SOD1-related ALS. Tofersen (marketed as Qalsody) is an antisense oligonucleotide designed to bind to SOD1 mRNA, ultimately reducing protein production. On April 25, 2023, the FDA announced the approval of tofersen for ALS patients demonstrating SOD1 mutation, making it the first treatment for SOD1-related ALS. This outcome was due to promising results of a 28-week, randomized, double-blind, placebo-controlled clinical trial of 147 patients demonstrating weakness and a confirmed SOD1 mutation. The patients receiving tofersen demonstrated significant reductions in plasma neurofilament light (NfL), a blood-based biomarker of axonal injury and neurodegeneration, which is reasonably likely to predict a benefit in the clinic. Before moving to clinical stages, this first-in-class therapeutic was evaluated using an in vivo model for SOD1-mediated ALS: the SOD1 rat.
Preclinical Support for Drug Development: The SOD1 Rat
The SOD1 rat is a hemizygous transgenic rat that overexpresses human SOD1 with the G93A mutation associated with familial ALS in the spinal cord as well as many other brain regions and peripheral tissues. The model was originally created by microinjection of a 12kb restriction fragment of the human SOD1 gene harboring the G93A mutation into Sprague Dawley rat embryos and multiple founder lines were generated. Founder line 26H was determined to have approximately 64 copies through qPCR, and a ratio of hSOD1/rSOD1 of 8.6 in spinal cord and 0.8 in blood by Western blot in juvenile rats. Lower copy number transgenic founders did not demonstrate disease pathology. The model first came to Taconic Biosciences in 2002 and has been maintained by breeding hemizygous carrier males to outbred commercial stock of female Sprague Dawley rats.
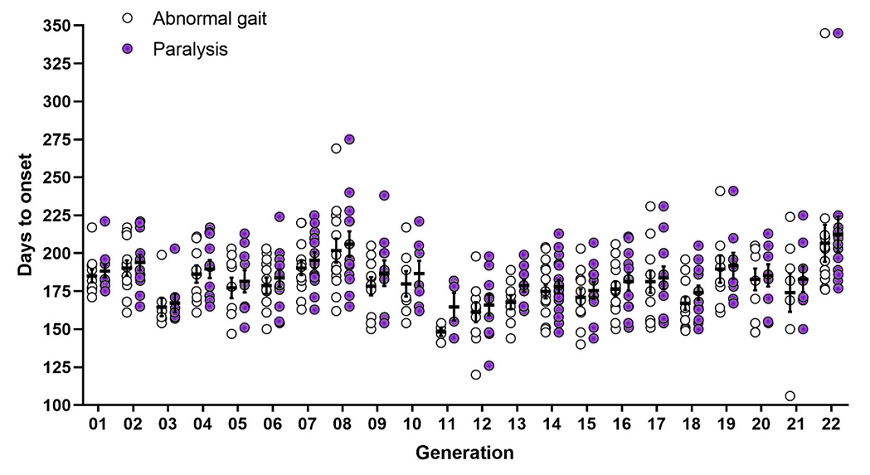
SOD1 rats from Taconic demonstrate onset of motor neuron disease with both intra- and inter-generational variability. Figure 1A below depicts the average timeframe for abnormal gait and paralysis onset in male SOD1 breeders maintained at Taconic for the past 29 generations, which varies considerably between generations. Abnormal gait is typically followed by hindlimb paralysis within 6-9 days. Figure 1B demonstrates the range of phenotype onset within each generation, which can also vary; for example, the average timeframe to abnormal gate in generation 22 was 223 days, but certain animals did not become symptomatic until 345 days.
{kind=link}
The SOD1 rat from Taconic was utilized by Ionis Pharmaceuticals in early development stages of tofersen. In the initial evaluation of two antisense oligonucleotides, both therapeutics were administered intrathecally into SOD1 rats. Rats on the treatments maintained weight up to 70 days longer than negative controls and had increased survival of up to 64 days compared to negative controls. These observations were statistically significant for both experimental therapeutics, and benefits were observed in both male and female mice. These promising findings warranted evaluation of the novel therapeutics in subsequent clinical trials5.
The ALS Association's Role in Promoting Access to Research Tools
To improve accessibility of this model to researchers, commercial production of the SOD1 rat at Taconic is sponsored by The ALS Association, the largest philanthropic funder of ALS research worldwide. The costs of production incurred by The ALS Association are offset by revenue generated via commercial sales. This sponsored colony approach of utilizing Taconic's rodent production and distribution expertise means labs around the world have been able to study the SOD1 rat for decades, resulting in advancements in the field and new treatments for people suffering from fatal neurodegenerative diseases. In addition to a multi-decade relationship with The ALS Association, Taconic also has a strong relationship with various other non-profit organizations and foundations focused on making rodent models available for research.
In 2022, the FDA released an action plan for advancing the development of safe and effective therapies for neurodegenerative diseases, including ALS. The plan acknowledged the lack of available treatments for people suffering from fatal disease and outlined tasks designed to bolster innovation, evaluation, and accessibility of novel therapeutics. Partnerships between Taconic, The ALS Association, and other non-profit organizations support this call for action by helping remove barriers to scientific discovery in neuroscience.